Stratification and sampling️ of population centers in Loreto, Peru 🎲✨
1 Introduction
This document describes the methodology carried out for the stratification and selection of population centers in Loreto for drone surveillance. This activity is part of the the Harmonize project: Harmonisation of spatio-temporal multi-scale data for health in climate change hotspots in the Peruvian Amazon (SIDISI 209821), in collaboration with the Barcelona Supercomputing Center.
The Harmonize project has the following objectives:
Geo-location of cases collected by passive surveillance from the Peruvian Ministry of Health (MINSA) through the Center for Disease Control and Prevention (CDC-Peru) in the study area using GPS devices.
Identify potential mosquito breeding sites based on new longitudinal ground survey data and using drone surveillance.
Collect data from weather stations, satellite imagery, ambient acoustic sounds and air pollution to calibrate and reduce the spatial resolution of Earth observation data sets.
Determine the impact of the use of these new technologies on infectious disease reduction interventions carried out by the Dirección Regional de Salud (DIRESA) of Loreto.
The methodology described here corresponds to the second objective above. Since we cannot map the whole study area of Loreto, our approach was to characterize the population centers in Loreto according to the incidence of relevant diseases in the area (Malaria, Dengue and Leptospirosis) and meteorological, environmental and human intervention variables and then use this variables to create sampling strata using clustering techniques. Finally, we randomly sampled a number of the population centers from each stratum proportional to the stratum size.
2 Dataset
For each population center, we gathered records on the following fields:
UBIGEOCP, which is the official identification code of the population centers used by the Instituto Nacional de Estadística e Informática (INEI).
Official name.
Latitude and longitude.
2022 cumulative incidence of Malaria, Dengue and Leptospirosis.
Difference in the cumulative incidence of Malaria between 2010 and 2019.
Mean and difference of the annual mean values of the years 2010 and 2021 of the following variables:
Precipitation accumulation (mm),
Runoff (mm),
Soil moisture (mm),
Maximum temperature (°C),
Minimum temperature (°C),
Total evapotranspiration (kg/m2),
Specific humidity (kg/kg),
Deforestation area (km2).
Mean and difference of the annual values of the years 2010 and 2020 of the following variables:
Population density from WorldPop and
Human settlement data from the Global Human Settlement Layer (GHSL).
Mean and difference of the annual mean values from years 2014 and 2021 of the nighttime data from the Visible Infrared Imaging Radiometer Suite (VIIRS).
We can take a look at the data in the following table:
The stratification of the population centers consisted of using the meteorological, environmental and human intervention variables to built groups of population centers with homogeneous characteristics. For this purpose, we tested two approaches. The first one consisted of performing an Agglomerative (“bottom-up”) Hierarchical Clustering (AHC) analysis using different distances, linkage methods, and number of clusters selection methods. On the second one, we used Principal Component Analysis (PCA) to reduce the dimension of the variables to one principal dimension and used it’s scores to create groups by quantile classification.
The AHC analysis consisted of two steps. First, we run several configurations the agglomerative nesting algorithms and selected the one which gave us the best hierarchical clustering (HC) structure. Secondly, from the selected hierarchical clustering structure, we calculated and examined a number of indices for determining the most appropriate number of clusters.
3.1.1 Chosing the hierarchical clustering structure
For this first step, we tested combinations of the following methods:
Scaling method: Whether to use normalization (scaling from 0 to 1) or standardadization (subtracting the mean and dividing by the standard deviation).
Distance: Whether to use euclidean distance or correlation-based distances, such as Pearson or Spearman correlation.
Linkage method: Whether to use average, single, complete, Ward or weighted average linkage method.
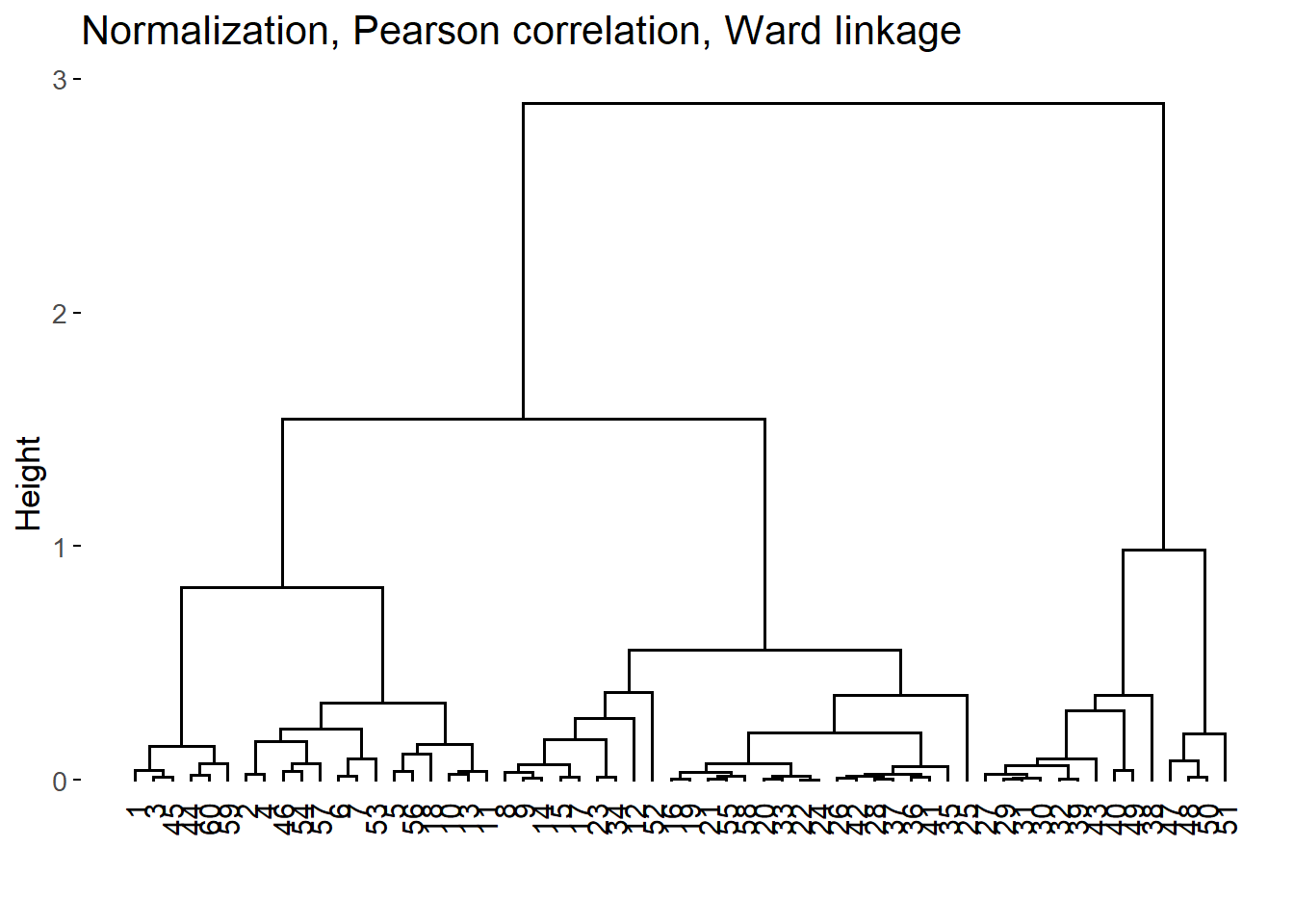
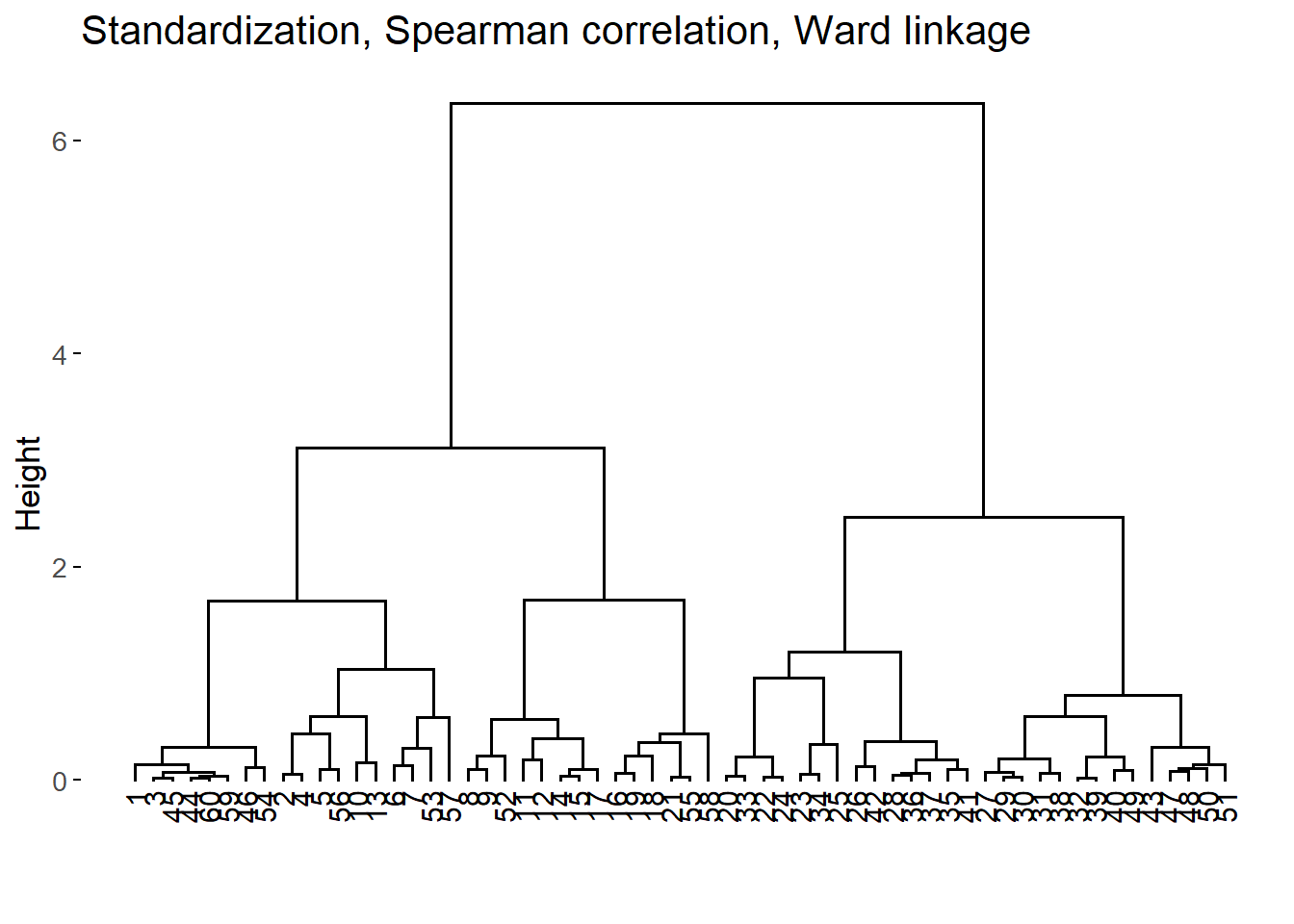
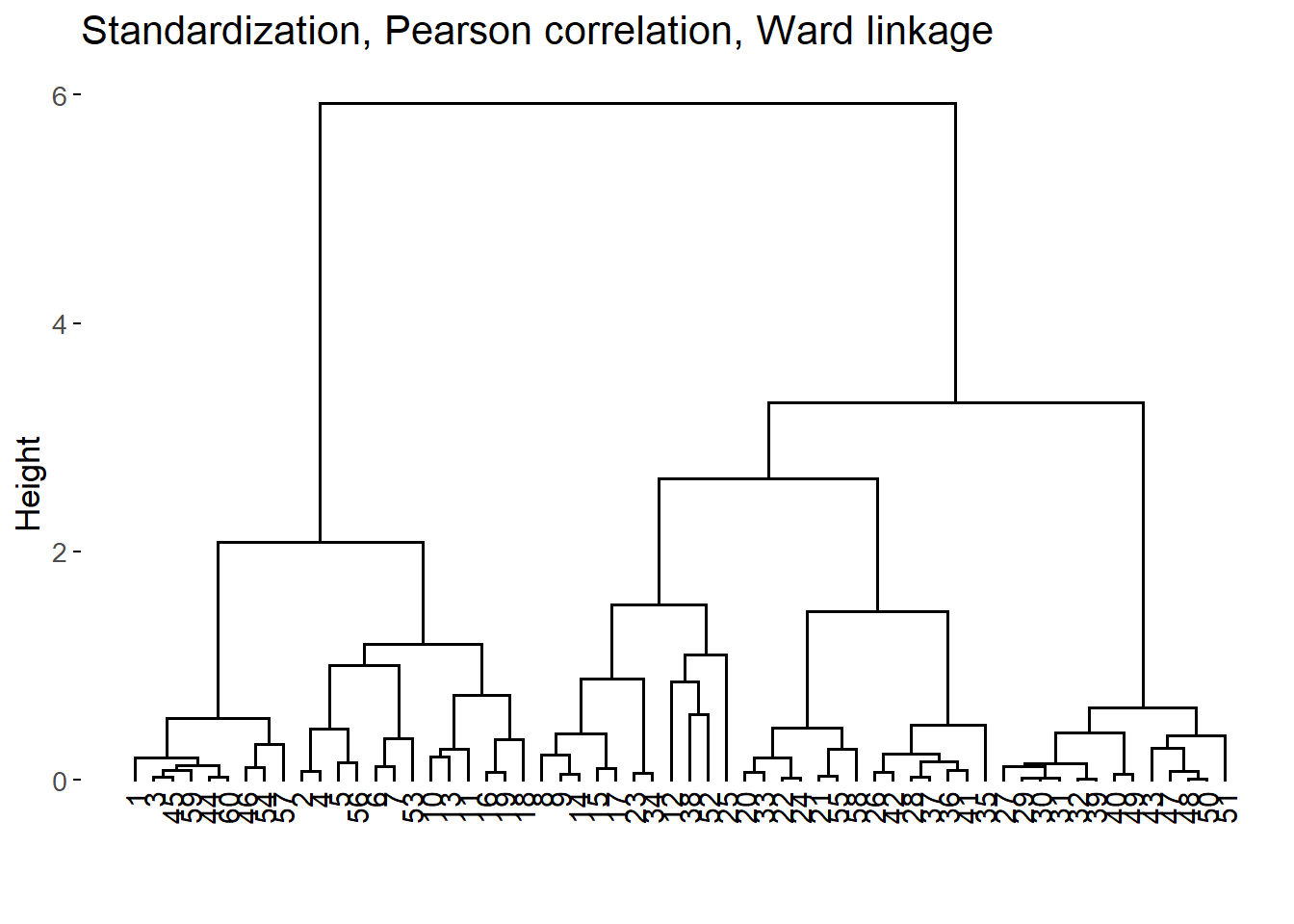
For each combination, we run the agglomerative nesting algorithm and calculated the agglomerative coefficient (AC) which is a measure of the strength of the HC structure (Boehmke and Greenwell 2019, 421–22). For the 3 combinations of methods with larger AC, we also examined the resulting dendograms.
Code
# Select variables for analysisvariables =select(ccpp, dengue:malaria_diff)# Function to scale data either by standardization or normalizationscale_data =function(data, method) {if (method =="standardization") { data_scaled =mutate(data, across(everything(), ~as.numeric(scale(.x)))) } if (method =="normalization") { data_scaled =mutate(data, across(everything(), scales::rescale)) } data_scaled}# Listing the configurations for the scaling, distance and linkage methodsscaling_method =c("standardization", "normalization")dist_method =c("euclidean", "pearson", "spearman")linkage_method =c("average", "single", "complete", "ward", "weighted")# Table with all the combinationsdesign = tidyr::expand_grid(scaling_method, dist_method, linkage_method, variables)# Calculate the agglomerative coefficient for each combinationagglomerative_coeff = design %>% tidyr::nest(data =-c(scaling_method, dist_method, linkage_method)) %>%mutate(scaling = purrr::map2(data, scaling_method, ~scale_data(.x, method = .y)),dist_mat = purrr::map2(scaling, dist_method, ~get_dist(.x, method = .y)),tree = purrr::map2(dist_mat, linkage_method, ~agnes(.x, method = .y)),ac = purrr::map(tree, ~.x$ac) ) %>% tidyr::unnest(ac) %>%arrange(-ac)
The best 3 HC structures according to the AC were the following:
The difference of the first 3 AC values are almost negligible. Consequently, we examined the dendograms:
Code
# Function to plot a dendogramdendogram =function(row, title) { dendogram <-fviz_dend(agglomerative_coeff$tree[[row]], main = title) +theme(text =element_text(size =13)) dendogram}
After inspecting the dendograms, we chose the second HC structure as it seems to yield more homogeneous groups with relatively similar sizes for the different height values.
3.2 Determining the number of clusters
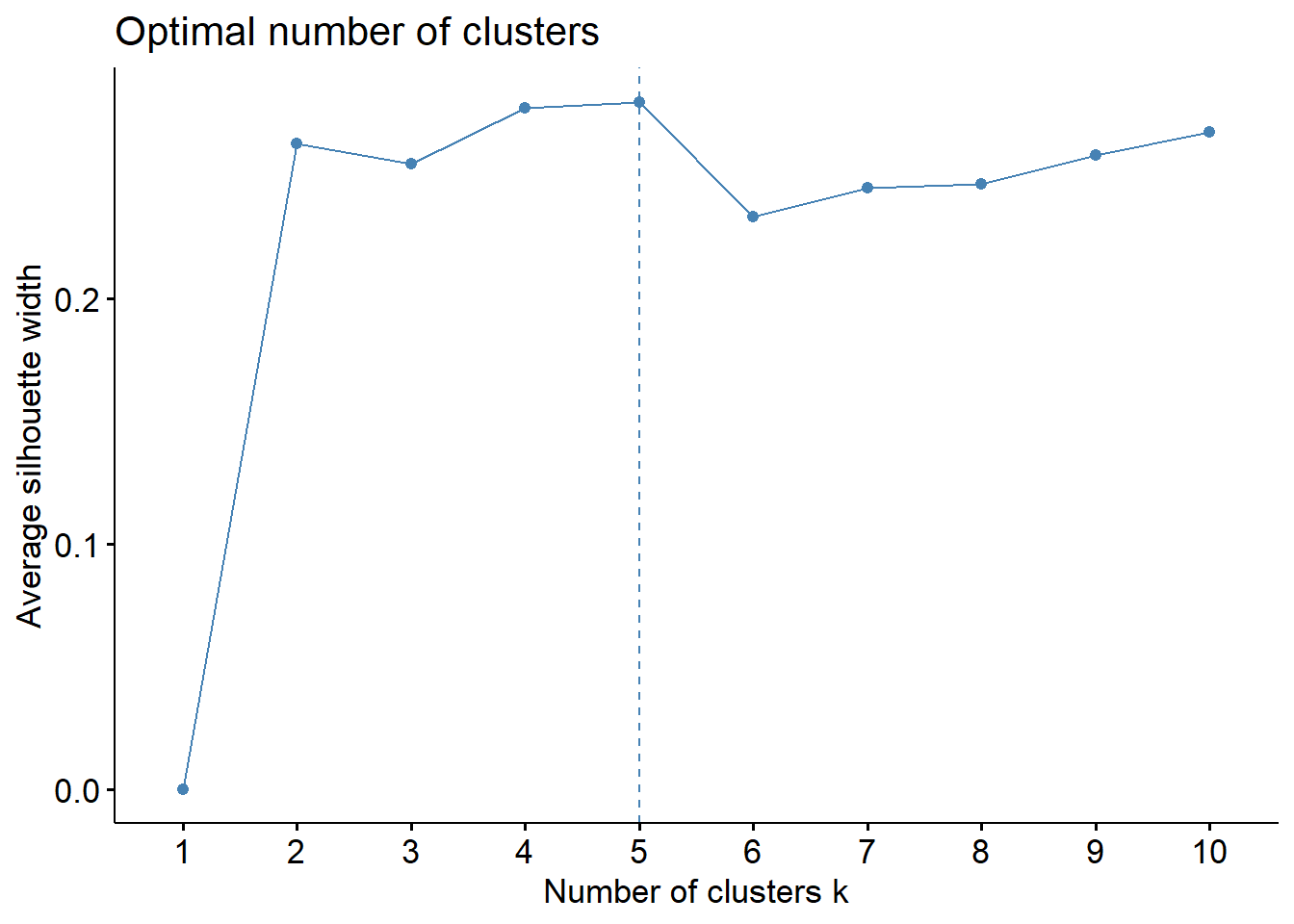
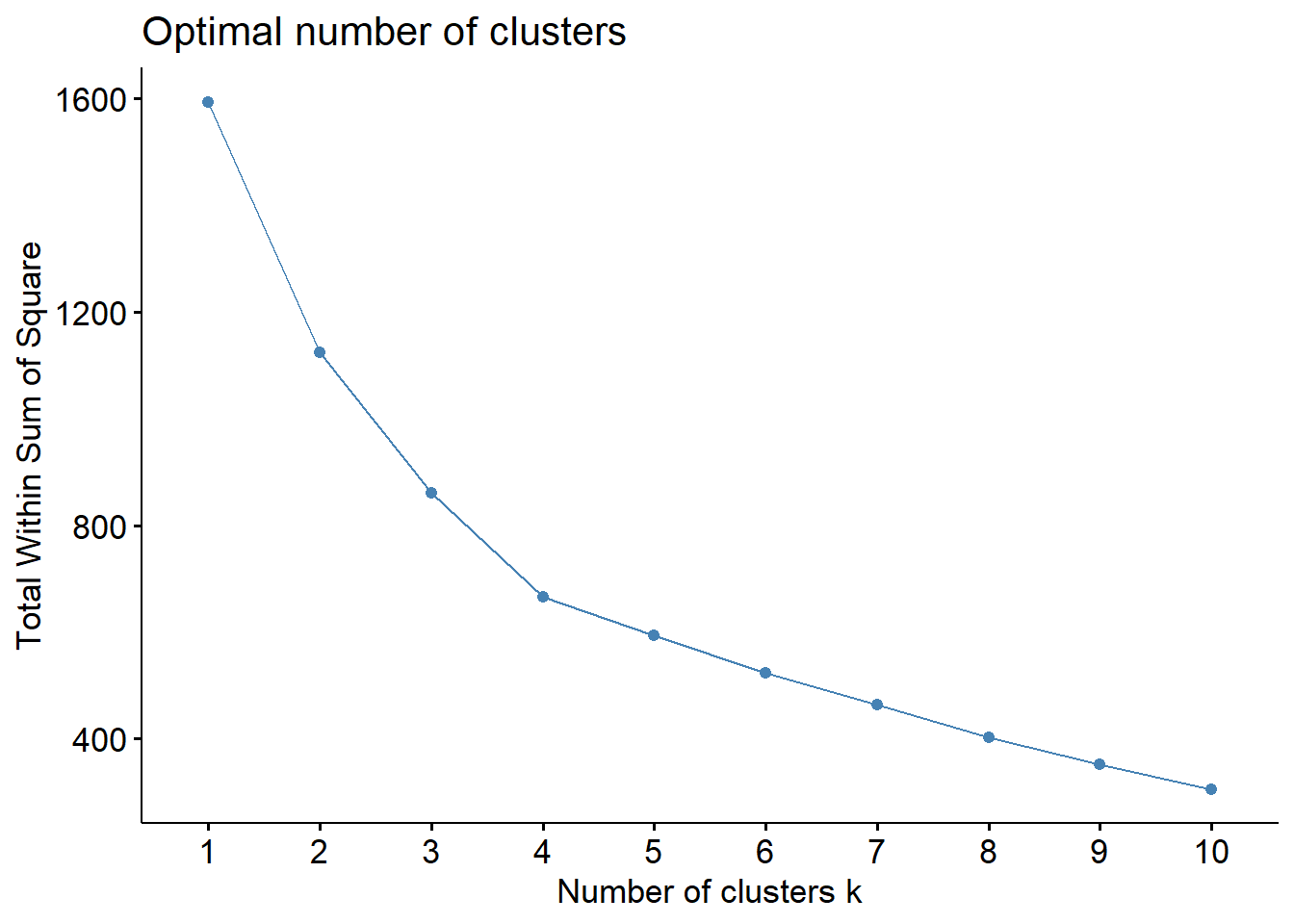
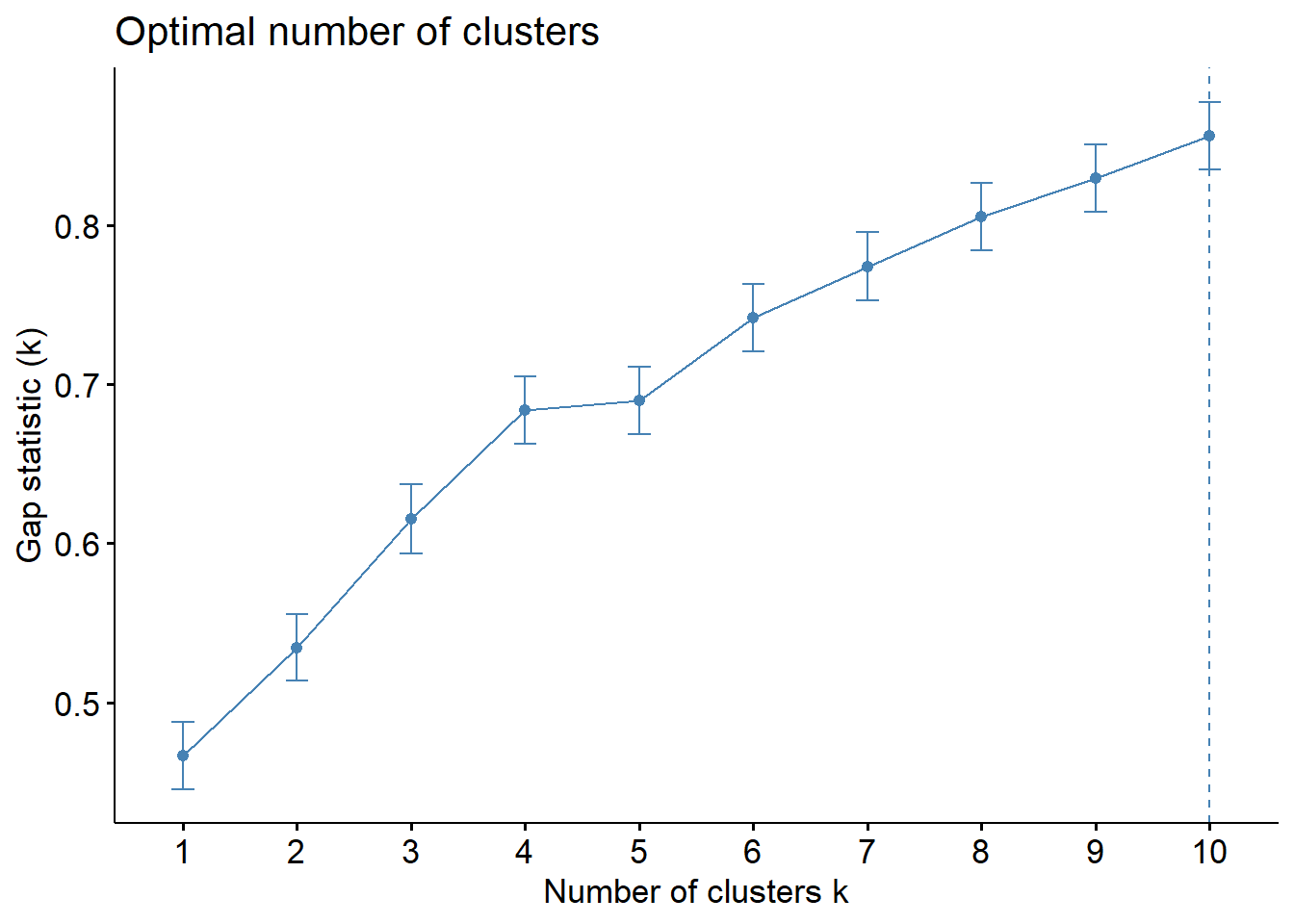
We calculated and plotted the Average silhouette width, Total within sum of squares and Gap statistic for a range of 1 to 10 number of clusters and examined the optimal number of cluster for each criteria. We defined a range of numbers of clusters that met or near met the optimal criteria and then for each one of them we used PCA to visualize the clusters in 2 dimensions to evaluate how well the groups were formed. In addition, the silhouette information of each observation was plotted to assess if there were observations that might have been misclustered.
3.2.1 Evaluation of the criteria of the optimal number of clusters
Code
# Function to plot optimal number of clusters plot for a certain criterianbclust_plot =function(data, method, ...) { nbclust <-fviz_nbclust( data, hcut, hc_func ="agnes", hc_method ="ward.D2", c_metric ="spearman", method = method, ... ) +theme(text =element_text(size =13)) nbclust}# Scale variables using standardizationvariables_std =mutate(variables, across(everything(), ~as.numeric(scale(.x))))
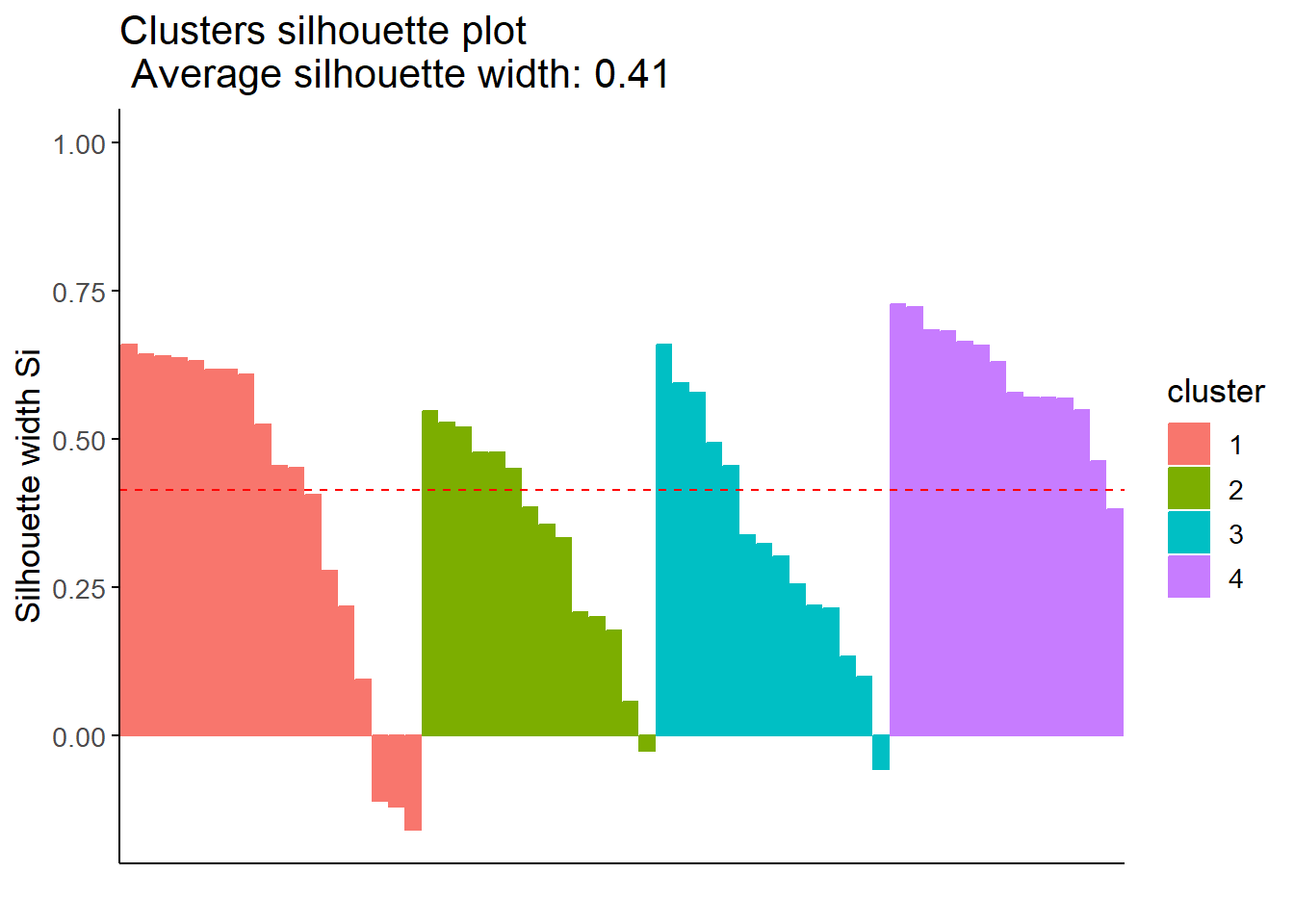
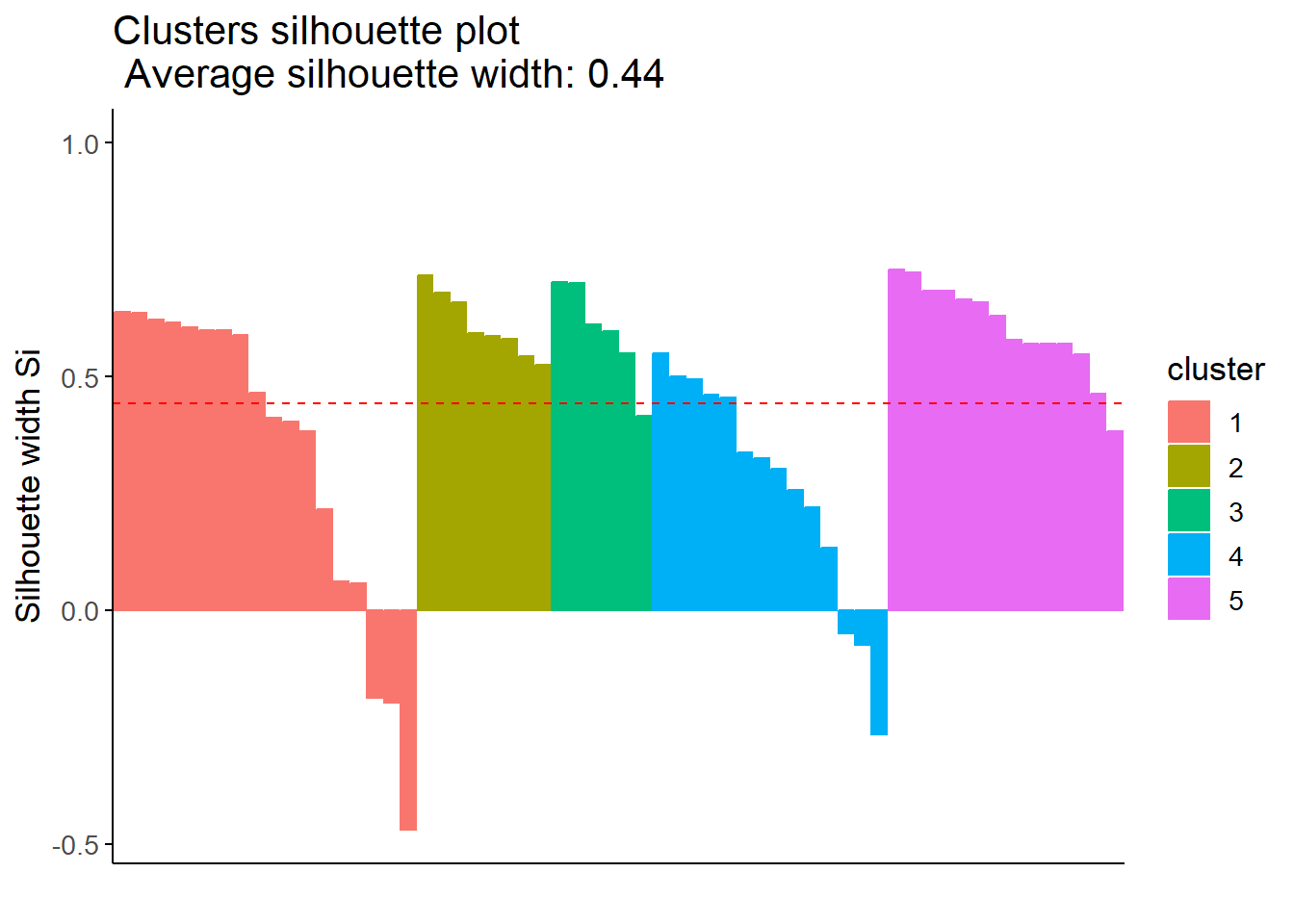
According to the average silhouette width, the optimal number of clusters is 5. In the total within sum of squares plot, we see an apparent elbow at 4 clusters. Finally, the Gap statistic tells us that the optimal number of clusters is 10 or more. However, we do not wanted to work with more than 5 clusters, 4 or 5 clusters would be the closest optimal.
3.2.2 Clusters plots
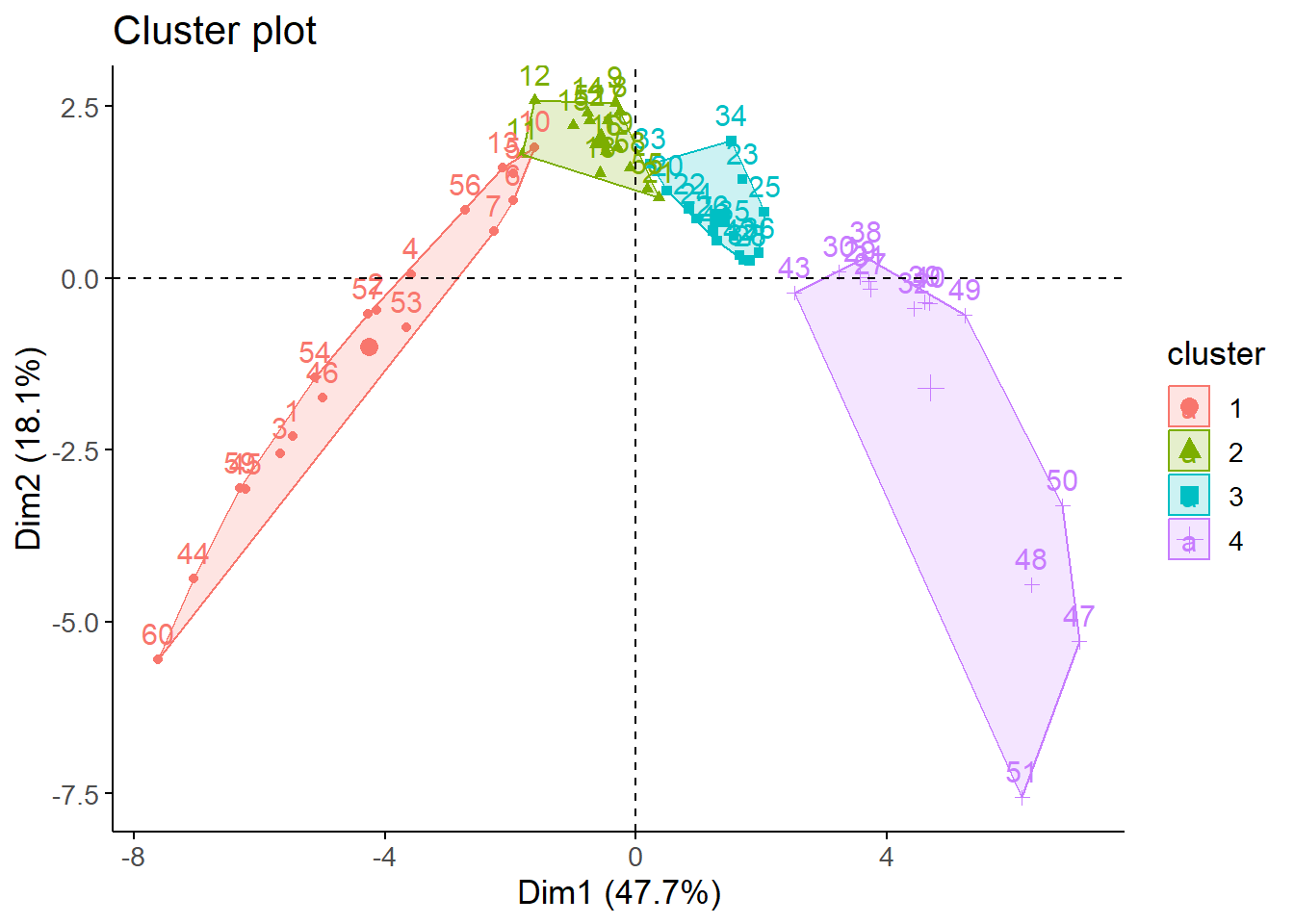
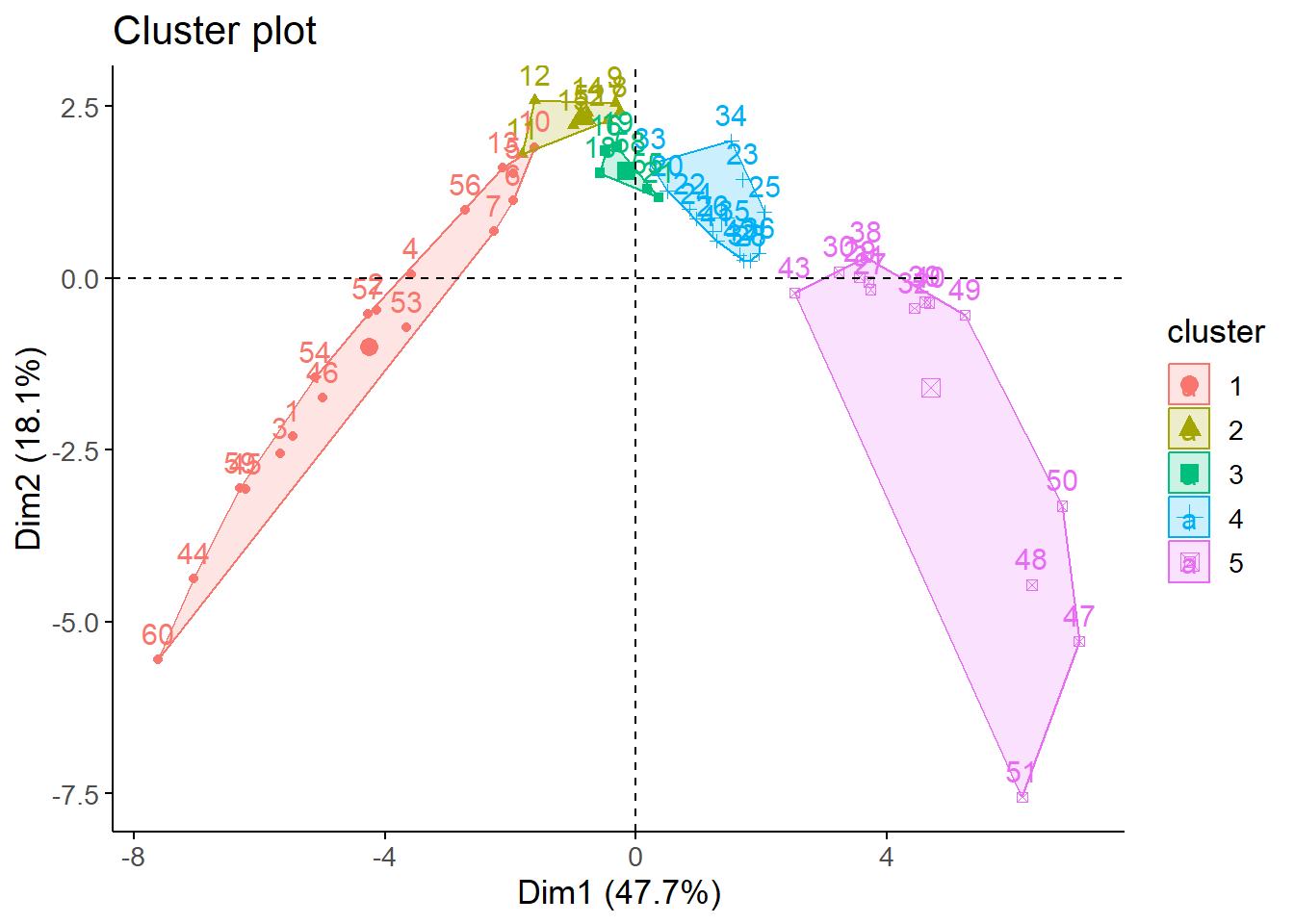
Next, we plotted the clusters in a plane using PCA for 4 and 5 clusters.
Code
# Function to cut treecut_tree =function(data, k) { tree =hcut( data, k = k, hc_func ="agnes", hc_method ="ward.D2", hc_metric ="spearman", graph =TRUE ) tree}# Function to generate cluster plotcluster_plot =function(tree) { cluster =fviz_cluster(tree, ggtheme =theme_classic()) +geom_vline(xintercept =0, linetype ="dashed") +geom_hline(yintercept =0, linetype ="dashed") +theme(text =element_text(size =13)) cluster}
We see that with 5 clusters we obtain a greater average silhouette width (0.44) than with 4 clusters (0.41). However, with 5 clusters with get greater negative values for some populated centers, indicating that they may be misclustered.
3.2.4 HC groups on maps
As an additional step, for each number of clusters, we plotted the population centers (polygon centroids) on a map to see their spatial distribution.
Code
# Create table with the groupsccpp_clusters = ccpp %>%mutate(hc_group_4 =as.factor(tree_4$cluster),hc_group_5 =as.factor(tree_5$cluster) )# Function to plot a map with the clusterscluster_map =function(data, palette, group_var) { values =pull(data, {{group_var}}) map = data %>% sf::st_as_sf(coords =c("lng", "lat"), crs =4326) %>%leaflet() %>%addTiles(group ="OpenStreetMap") %>%addProviderTiles(provider = providers$CartoDB, group ="CartoDB") %>%addProviderTiles(provider = providers$Esri.WorldImagery, group ="Satelital" ) %>%addCircleMarkers(popup = ccpp$population_center, color =~palette(values),opacity =1, radius =0.1, fillOpacity =0.5 )%>%addLayersControl(baseGroups =c("CartoDB", "OpenStreetMap", "Satelital") ) %>%addLegend(title ="Cluster", pal = palette, values = values, opacity =1,position ="bottomright" ) map}
We see that the population centers seem to group, showing a strong spatial component driving the clustering.
3.3 Principal components analysis
Principal component analysis (PCA) was used as an alternative method for clustering. Our approach was to use the principal component to build an aggregate index of all the variables considered. Then, the scores of the populated centers on this principal component were grouped into different categories according to the quantiles of the distribution. In order to compare this approach with the results of hierarchical clustering, we tested with 4 (25th, 50th and 75th quantiles) and 5 (20th, 40th, 60th and 80th quantiles) groups.
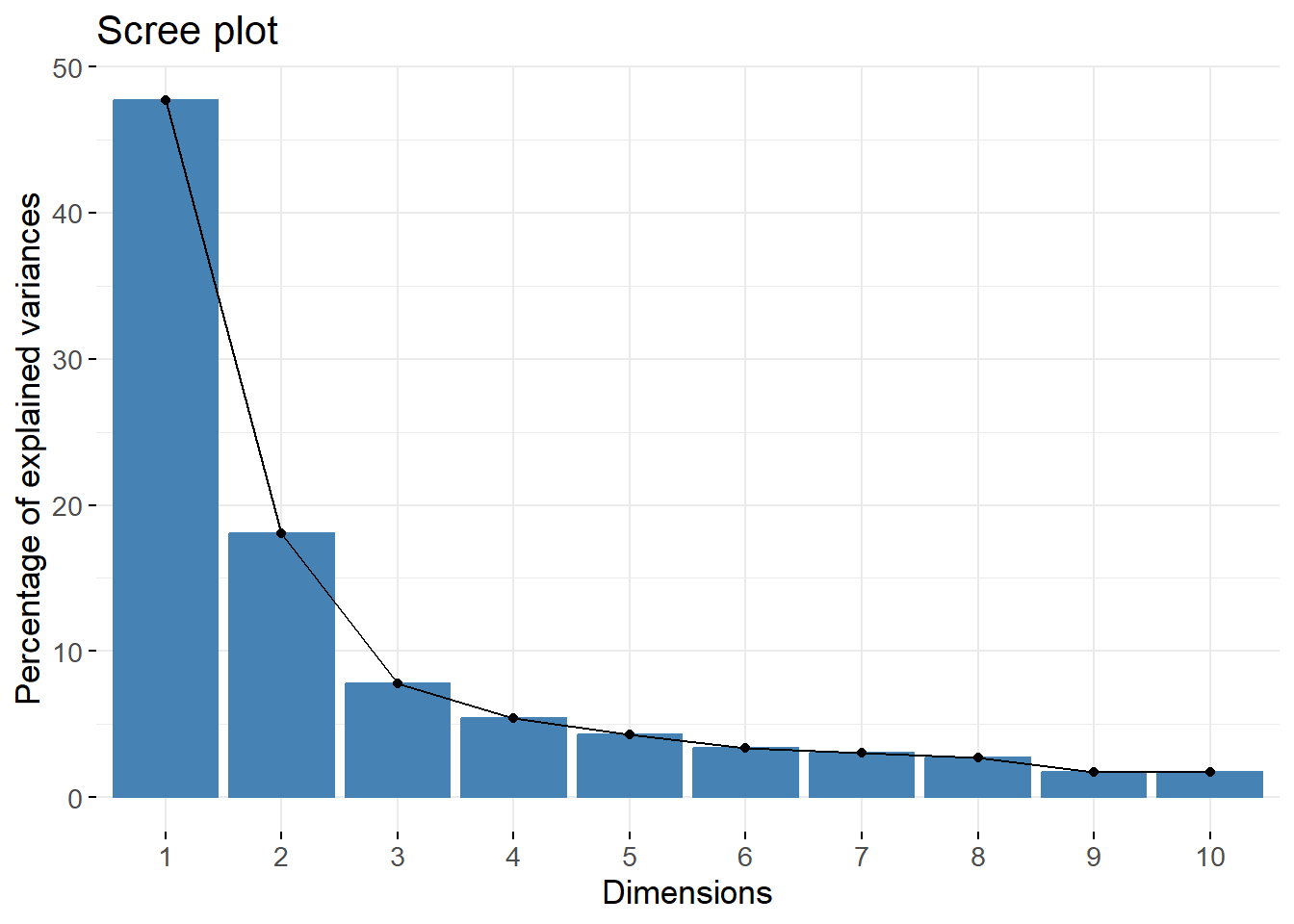
We see that the first component explain approximately 50% of the variance in the data. This is not such a high value, meaning that the variables do not seem to be strongly linearly correlated. Still, 50% is an acceptable value in practice.
3.3.2 Contribution plot
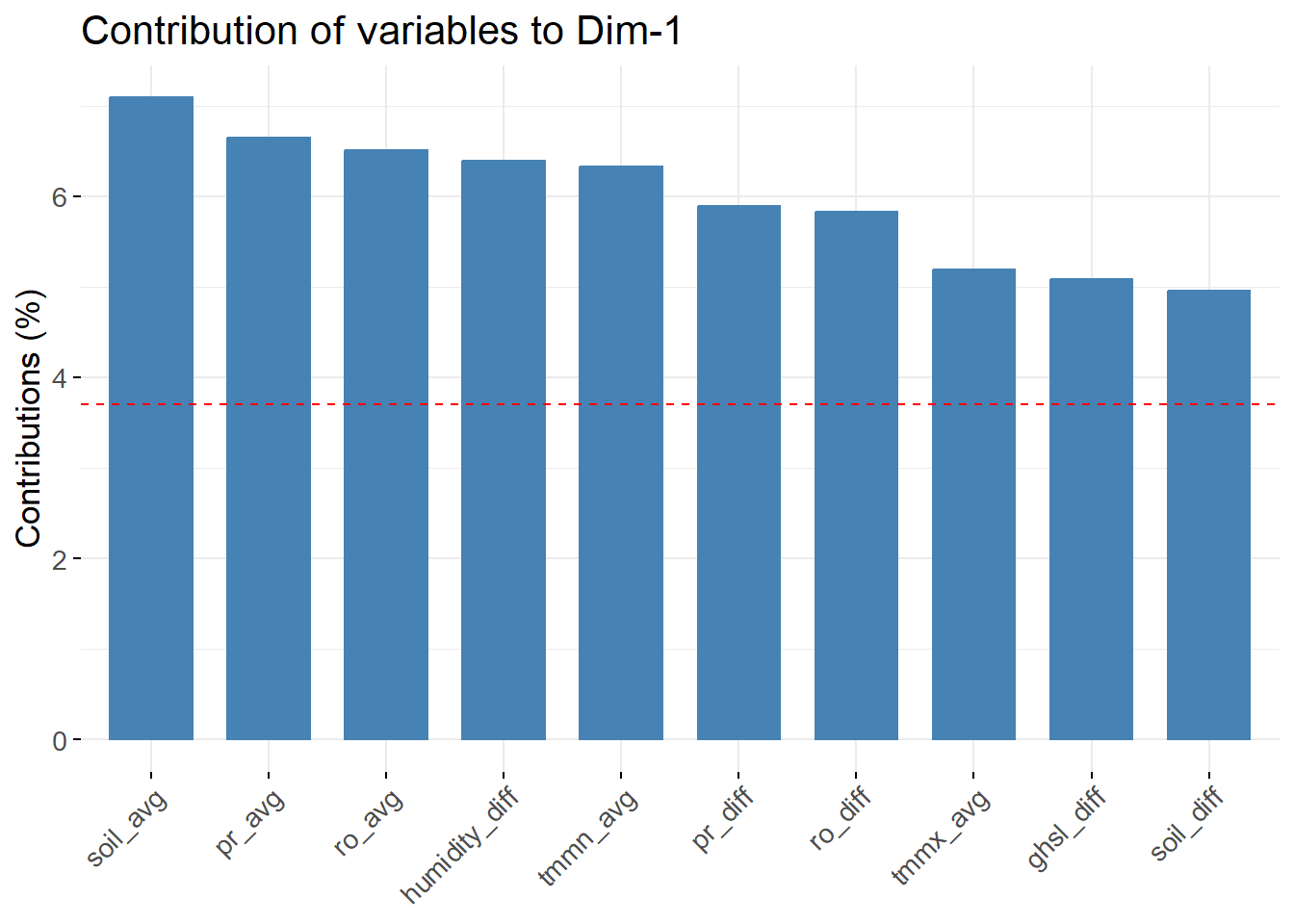
Next, we plotted the top 10 variables according to their contribution to the first component.
Code
pca %>%fviz_contrib(choice ="var", axes =1, top =10) +theme(text =element_text(size =13))
We see that the soil moisture, precipitation and runoff variables are the ones that contribute the most to explaining the first component.
3.3.3 PCA groups on maps
Finally, we created the groups dividing the population centers according to the quantiles of the scores on the principal component. Then we plotted the population centers on maps for 2, 3 and 4 groups.
Geographically, the PCA groups seem to be distributed very similar to the HC groups.
3.4 Comparing HC and PCA
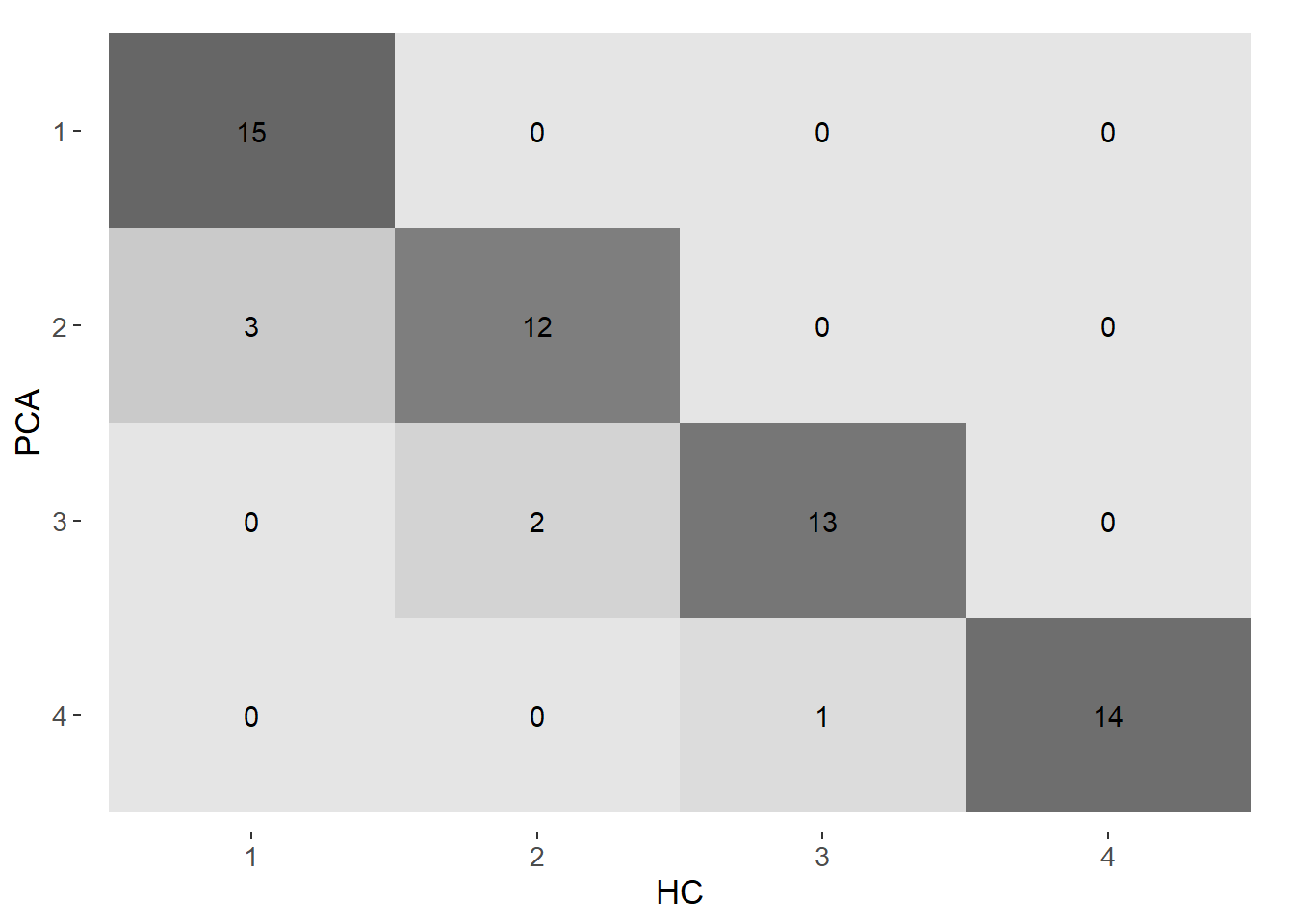
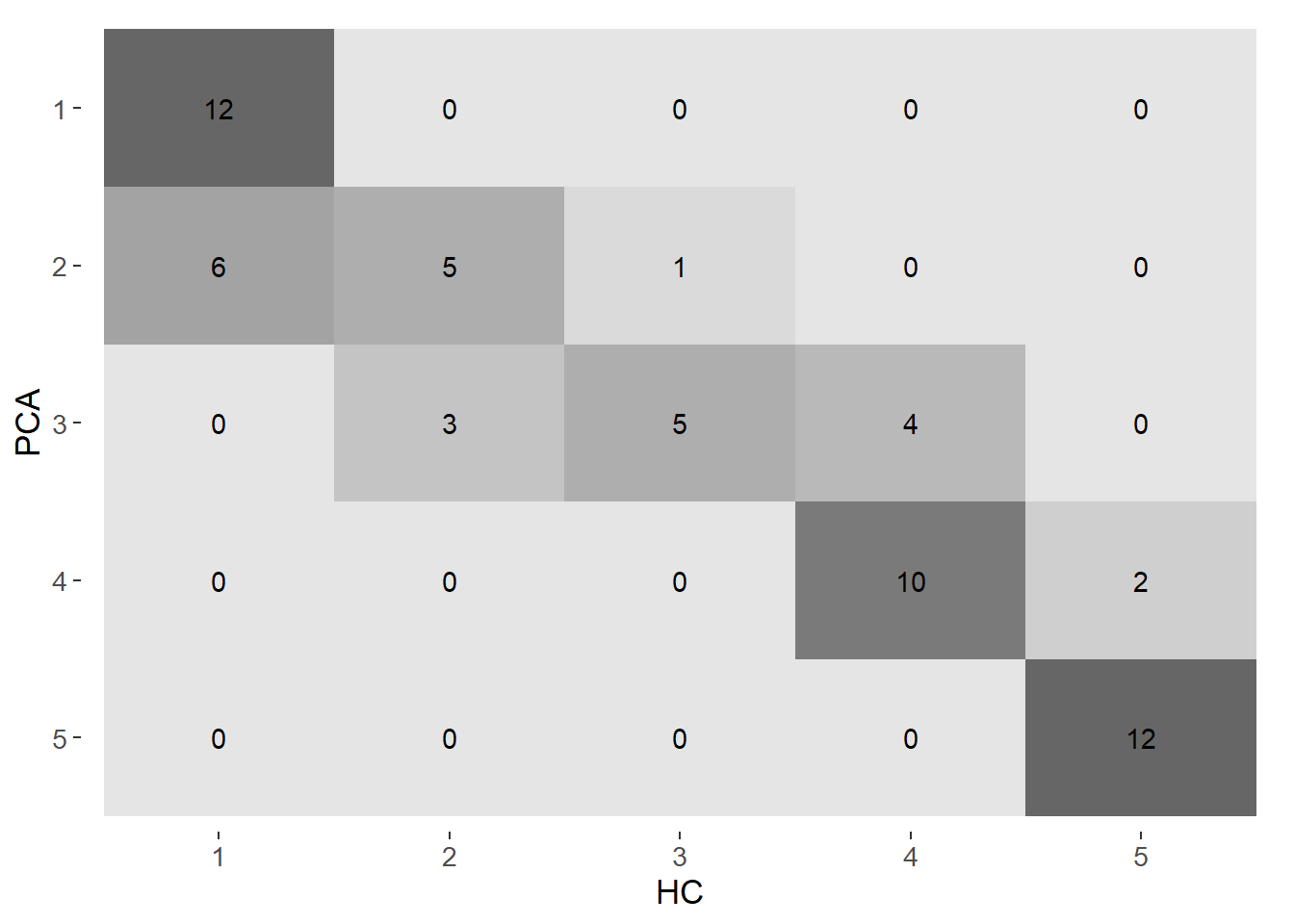
We assessed how strong was the agreement between the HC method and PCA method on the grouping of the population centers for 4 and 5 groups. Confusion matrices and the Kappa statistic were used for this analysis.
From the confusion matrices it is evident a moderate agreement between the HC method and the PCA method. The agreement is stronger with 4 groups (kappa = 0.87) than with and 5 (kappa = 0.67).
In summary, both 4 and 5 groups give very similar results in terms of visual and statistical indicators. However, we ultimately opted to work with 4 clusters to avoid an excessive disaggreation of populated centers.
4 Selection of populated centers
Having chosen the number of clusters, we determined the number of population centers we should select from each one of them to obtain a sample of 10 population centers with the same proportion of cases in each cluster.
Code
# Sample size for each clusterround(table(ccpp_strata$hc_group_4)/6)
1 2 3 4
3 2 2 2
Therefore, we needed to select roughly 3 populated centers from cluster 1 and 2 from each of the other ones.
We did not take a random sample approach since we had the following restraints:
The Direccion Regional de Salud de Loreto (DIRESA Loreto) selected 5 population centers that are of epidemiological interest.
We had previously conducted field work to collect georeferenced points to reconstruct the perimeter of 21 populated centers. This is work that would have to be done for all the communities selected for drone mapping. So, for the sake of convenience, it was better to select from these population centers whose perimeters we already had.
We also filtered out those communities that required more than 3 hexagons of 1km2 area for drone flights.
In the end, we were left with 24 populated centers (5 from DIRESA Loreto, 19 from InnovaLab), and we took these as our sampling frame. The 5 populated centers from DIRESA Loreto were a must and 4 of them were in cluster 1 and 1 in cluster 2, so from the InnovaLab collection we randomly sample 1 populated center from cluster 2 and 2 from clusters 3 and 4 to have a total sample size of 10.
The size and the percentage from the total of the clusters and the final sample are shown in the following table: